Introducción
La enfermedad de Von Hippel Lindau es una enfermedad hereditaria, autosómica dominante, ocasionada por la mutación de un gen, y que predispone al portador de la misma al desarrollo de tumores en diferentes órganos. Entre ellos destacan : hemangioblastomas en el SNC, angiomas retinianos, quistes y cáncer renal, feocromocitomas, quistes y tumores pancreáticos, tumores de saco endolinfático (ELST) y quistes en epidídimo y ligamento ancho. La mayoría de los tumores son multicéntricos o bilaterales, y aparecen a edades más tempranas que en los no portadores de la mutación.
Los avances en citogenética de los últimos años han demostrado que el cáncer es una enfermedad genética. La mutación cromosómica que más frecuentemente se ha visto asociada a la enfermedad, se encuentra en el cromosoma 3, en la posición 3p25- 26, provocando la desaparición de un gen supresor de tumores, y facilitando así que la respuesta del sistema inmune ante la aparición natural de mutaciones celulares se vea mermada. Sin embargo, en muchos pacientes diagnosticados clínicamente, la mutación cromosómica que permitió la aparición de la enfermedad permanece desconocida durante años.
La incidencia aproximada de la enfermedad de Von Hippel Lindau está en 1/36.000. Se sospecha que es incluso más frecuente pero, debido al desconocimiento de la misma, se cree que está infradiagnosticada.
La penetrancia es alta en los portadores de la mutación, de forma que casi todos los portadores han desarrollado tumores relacionados con la mutación del gen a la edad de 60 años. Las primeras manifestaciones de la enfermedad pueden darse a cualquier edad, y habitualmente son síntomas neurológicos o defectos del campo visual (escotomas). A nivel cerebeloso, cuando el tumor ha alcanzado un tamaño que compromete el tejido circundante, comienza la sintomatología, consistente en cefaleas diarias, náuseas, vómitos, mareos y alteraciones del equilibrio evidenciables en la exploración neurológica. Los angiomas retinianos llegan a alcanzar gran tamaño, produciendo desprendimientos de retina y en ocasiones hemorragias, con la consiguiente pérdida de visión. En cualquier caso, estos angiomas aun pequeños, van disminuyendo la visión conforme crecen, por el exudado circundante que provocan.
El cáncer renal aparece alrededor de los 40 años. En muchas ocasiones es bilateral y causa importante de mortalidad. El gen marcador de la enfermedad se ha encontrado mutado en tumores esporádicos de pacientes sin VHL, especialmente en el carcinoma renal de células claras (hasta en el 85% de los cánceres renales estudiados por el Instituto Nacional del Cáncer (NCI) estadounidense (Duan y colaboradores, 1995). Se aconseja estudio genético en todo paciente joven diagnosticado de un tumor renal, por la posible asociación con la enfermedad de VHL. Actualmente las metástasis del carcinoma renal y las complicaciones neurológicas de los hemangioblastomas cerebelosos son las causas más comunes de muerte. Sólo el seguimiento adecuado y la cirugía a tiempo están contribuyendo a disminuir la morbilidad y la mortalidad.
Los pacientes que tengan un tumor descrito asociado a la enfermedad y una historia familiar negativa, pueden ser portadores de una mutación. Esto es posible por:
- 1) una mutación de novo en la línea germinal materna o paterna.
- 2) mosaicismo somático – de forma una parte de las células del individuo portan la mutación, y otras no- , debido a que se produjera ésta durante la embriogénesis.
- 3) La enfermedad no se ha expresado en los padres.
El estudio del DNA y la identificación de los portadores para seguimiento desde temprana edad se perfilan como la mejor prevención de lascomplicaciones. Aquellos miembros de la familia que, tras el estudio genético, se compruebe que no son portadores de la mutación, no precisan someterse a ningún otro tipo de pruebas. La gran dificultad para el diagnóstico surge cuando el paciente es el primer caso dentro de la familia (mutación â??de novoâ?).
Una vez hecho el diagnóstico, el paciente ha de seguir controles periódicos rigurosos durante toda su vida, pues el pronóstico depende en gran medida de la detección temprana de los tumores. Anualmente se efectúa resonancia magnética nuclear (RMN) para el seguimiento de los tumores cerebelosos. Los exámenes visuales se programan cada seis meses, pues la detección de los angiomas cuando todavía son pequeños permite el tratamiento con láser. Cada seis meses se realiza una ecografía abdominal para control de páncreas, riñones y glándulas suprarrenales, que se completará con TAC en caso de que existan dudas diagnósticas. Se recomienda determinación de catecolaminas en orina de 24 horas, anualmente, para despistaje de feocromocitomas.
No hay estudios suficientes para hacer recomendaciones generales, pero parece que el embarazo no tiene efecto sobre la enfermedad. Antes de un embarazo, la paciente ha de someterse a un chequeo riguroso debido a las posibles complicaciones que pudieran representar tumores no detectados previamente.
La esperanza media de vida se ha estimado en unos 49 años. Sin embargo, los datos de la historia natural de la enfermedad son escasos, por lo que la esperanza de vida de pacientes no diagnosticados podría ser mucho menor.
Debe comenzarse de inmediato el estudio del árbol genealógico familiar. Si se ha conseguido determinar la alteración cromosómica responsable de la enfermedad en la familia, el estudio se inicia con el análisis genético de los padres. La enfermedad no tiene una correlación clara genotipoâ??fenotipo, por lo que se siguen estudiando las variantes cromosómicas en relación con las características fenotípicas de los pacientes.
Las investigaciones más recientes han propiciado el descubrimiento de fármacos inhibidores de la angiogénesis, que están siendo objeto de ensayos clínicos en la actualidad. Su mecanismo de acción se basa en la supresión de la señal intracelular que induce la producción excesiva de factor de crecimiento endotelial vascular (VEGF).
Características clínicas y manejo de la enfermedad de VHL.
1. Manifestaciones oculares.
Los angiomas retinianos son los más frecuentes y tempranos de los tumores que aparecen en esta enfermedad. Hasta en un 43 % de los casos son la primera manifestación. La mayoría son periféricos (alrededor de un 90%), un 5% se encuentran en la papila del nervio óptico, y un 5% en la mácula (los porcentajes varían ligeramente según las series estudiadas).
En el examen histopatológico se observa que están compuestos de una proliferación de capilares, y células estromales y gliales, idénticas al hemangioblastoma cerebeloso.
Los hemangioblastomas de la retina periférica habitualmente se hacen sintomáticos durante la tercera década de la vida. Pueden causar disminución de la agudeza visual, o alteraciones del campo visual debidas al exudado que provoca el tumor, hemorragias en la vecindad del mismo, desprendimientos de retina o pliegues en la mácula. En estudios de seguimiento se ha constatado la lenta progresión de estos tumores (los cambios objetivados por el observador no fueron significativos a lo largo de 4-5 años). Puede ocurrir regresión espontánea, aunque no es lo habitual.
Recientemente, Webster et al. estudiaron la historia natural de 183 pacientes de 81 familias. La media de tumores en los portadores de la mutación era de 1,85 y variaba entre 1 y 15 lesiones. La probabilidad acumulativa de pérdida visual a la edad de 50 años fue de 35% en los pacientes portadores, 55% en aquellos diagnosticados de tumores retinianos, y significativamente peor en pacientes con lesiones sintomáticas. El 68% de los pacientes VHL estudiados tenía hemangioblastomas en retina, y los tumores se diagnosticaron a una edad media de 31 años (rango: 7 a 74 años). Los investigadores observaron que la prevalencia de tumores retinianos no aumentaba con la edad.
El diagnóstico se realiza por examen oftalmológico, midiendo la agudeza visual, examen con lámpara de hendidura y oftalmoscopia (con dilatación pupilar completa). Si los hemangioblastomas están localizados en la retina periférica, se debe realizar la inspección con lente de tres espejos Goldmann. En ocasiones se indica un estudio angiográfico con fluoresceína para poner de manifiesto lesiones muy pequeñas.
Se han probado diferentes tratamientos para este tipo de tumores: fotocoagulación con láser, criocoagulación, interferón α 2a (reduce el tamaño de los mismos) y braquiterapia. Todos han sido efectivos dependiendo de la localización, tamaño y número de los tumores. En el caso de los angiomas localizados en las proximidades de la mácula o en el nervio óptico, no es aconsejable el tratamiento debido al riesgo de pérdida visual, por lo que sólo cabe la conducta expectante. Cuando los tumores van creciendo, traccionan de la retina ocasionando desprendimientos y hemorragias, lo que hace inevitable la intervención quirúrgica. En estos estadios es frecuente la aparición de glaucoma secundario, acompañando a la pérdida completa o casi completa de visión. En muchos de estos casos es precisa la enucleación para suprimir el dolor.
Las decisiones son siempre difíciles y muchas veces sólo está justificado un seguimiento sin tratamiento.
2. Sistema nervioso central.
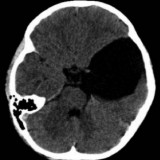
Los hemangioblastomas del SNC son la causa del diagnóstico de la enfermedad en el 40% de los pacientes aproximadamente. Se localizan predominantemente en el cerebelo, aunque también pueden aparecer en la médula (intradurales) y tronco cerebral. Más raramente se han observado en los hemisferios cerebrales. Pueden ser únicos o múltiples (se han descrito casos de pacientes con más de 30 tumores, que han ido surgiendo a lo largo de los años). La edad media del diagnóstico es de 30 años.
Al igual que los angiomas retinianos, son benignos, y de crecimiento lento. Anatomopatológicamente están compuestos predominantemente de células estromales y vasculares. Se han clasificado macroscópicamente en cuatro tipos: quísticos (5%), predominantemente quísticos (60%), predominantemente sólidos (26%) y sólidos (9%).
La sintomatología depende de la ubicación y tamaño de los tumores. Los síntomas más comunes en el momento del diagnóstico son cefaleas, náuseas, vómitos, mareos, ataxia y sensación de inestabilidad. En el caso de los tumores medulares, el dolor es el síntoma más frecuentemente referido.
El diagnóstico se realiza mediante RMN con gadolinio. Hasta el momento, el tratamiento definitivo de los tumores sintomáticos es la cirugía, complementada en caso necesario con una embolización previa del tumor. El pronóstico ha mejorado notablemente con el desarrollo de la microcirugía y los avances en cuidados intensivos. Sin embargo, la posibilidad de desarrollo de nuevos tumores todavía es un grave problema.
Gracias a los programas de seguimiento de la enfermedad que se llevan a cabo, muchos tumores son detectados en fase asintomática. Hay que sopesar muy cuidadosamente el riesgo de la intervención frente a la evolución natural del tumor, pues en este último caso la complicación aguda más temida en tumores asintomáticos â??la hemorragia- aparece con una frecuencia muy baja (0-1%).
En los últimos años se ha comenzado a utilizar una nueva técnica para el tratamiento de estos tumores, la radiocirugía estereotáctica. Esencialmente consiste en bombardear el tumor con haces de radiación desde diferentes ángulos. El tejido sano circundante recibe una dosis muy pequeña, mientras que en la lesión confluyen las radiaciones individuales. Ofrece la posibilidad de tratar varios tumores en una sola sesión. En estudios realizados hasta la fecha se ha constatado que la radiocirugía reduce o detiene el crecimiento de tumores sólidos (se utiliza en aquellos menores de 3 cm), pero los quistes asociados no responden, por lo que no está indicado su uso en estos casos. Actualmente se está utilizando cada vez más esta técnica para el tratamiento de los hemangioblastomas del SNC en la enfermedad de von Hippel Lindau, pero se requieren estudios a largo plazo, para determinar tanto resultados como complicaciones.
Todos los tipos de mutaciones del gen VHL predisponen para el desarrollo de hemangioblastomas del SNC.
3. Cáncer renal y quistes.
Las lesiones renales aparecen de forma uni o bilateral, únicas o múltiples, aunque lo más frecuente es que sean múltiples y bilaterales. Mientras que el cáncer renal esporádico aparece por encima de los 60 años, en estos pacientes se presenta con una media de edad de 36 años. Los quistes renales habitualmente son asintomáticos, mientras que el cáncer renal se puede presentar con dolor lumbar y hematuria. El diagnóstico en estadios presintomáticos es esencial para la supervivencia del paciente. Histológicamente el patrón celular más común de cáncer renal en esta enfermedad es el carcinoma de células claras. Presenta una serie de características diferenciales con los tumores esporádicos que aparecen en personas sin VHL: habitualmente se rodean de una cápsula de tejido conectivo, el pronóstico es significativamente mejor que en aquellos pacientes no VHL, y el crecimiento es más lento que en dichos tumores esporádicos. El estudio anatomopatológico de las lesiones ha revelado que la mutación del gen se encuentra tanto en los quistes como en el carcinoma, por lo que se piensa que aquellos son los precursores del cáncer (no está del todo claro).
Algunos protocolos de seguimiento recomiendan ecografía cada seis meses, y TAC cada dos años, para reducir tanto los costes como la exposición a la radiación. Otros protocolos combinan los ultrasonidos con la RMN abdominal.
El tratamiento depende de las características de los tumores, variando desde el seguimiento cuidadoso de las lesiones a la nefrectomía bilateral. Actualmente se tiende a preservar al máximo la función del órgano gracias a la microcirugía.
El tamaño del tumor tiene gran importancia en las decisiones terapéuticas. Se ha establecido como límite los 3 cm de diámetro para decidir intervenir al paciente, pues se ha observado que por debajo de este tamaño no suelen metastatizar. Los enfermos de VHL en los que se ha efectuado nefrectomía bilateral, son buenos candidatos a trasplante, pues el riñón donado no tiene la mutación, por lo que no tiene porqué desarrollar un nuevo cáncer.
Todos los tipos de mutación del gen VHL predisponen al cáncer renal, que es una de las principales causas de muerte en esta enfermedad.
4. Feocromocitomas.
El feocromocitoma puede ser la única manifestación de la enfermedad, aun habiendo detectado la mutación mediante análisis del DNA. Este tumor, secretor de catecolaminas, se origina a partir de células cromafines de la cresta neural, por lo que los tumores (paragangliomas) se pueden encontrar en tejidos cromafines del cuello, cavidad torácica, cavidad abdominal e incluso intravesicales. El término feocromocitoma (paraganglioma medular) está oficialmente reservado para designar a los tumores de la médula adrenal, aunque en muchas ocasiones encontraremos el término â??feocromocitoma ectópicoâ? para hacer referencia a los feocromocitomas extra- adrenales. Menos del 1% sigue un curso evolutivo maligno.
Hay una clara correlación entre el tipo de mutación (la denominada â??Missense Mutationâ?) y la aparición de feocromocitomas en la enfermedad de VHL. La edad media del diagnóstico es de 28 años, aunque el paciente más joven diagnosticado tenía 5 años.
La mayor parte de los feocromocitomas contiene y secreta tanto noradrenalina como adrenalina, pero la mayoría de los extra-adrenales secretan sólo noradrenalina. La sintomatología que encontraremos se debe a la liberación de catecolaminas a la circulación general. Los síntomas más frecuentes son: cefaleas, sudoración, palpitaciones con o sin taquicardia, nerviosismo, pérdida de peso, dolor abdominal o torácico, náuseas con o sin vómitos, astenia, ataques de pánico, hipertensión mantenida, crisis hipertensivas, etc.
En los pacientes con VHL el feocromocitoma a menudo permanece quiescente durante mucho tiempo, o produce mínimos síntomas. Los análisis bioquímicos pueden ser normales. Su evolución es imprevisible.
El diagnóstico se basa en estudios bioquímicos y radiológicos. La valoración de normetanefrina (catabolito de la noradrenalina) en plasma es el más sensible (97%), pero habitualmente se realiza como screening la detección de catecolaminas en orina de 24 horas (adrenalina, noradrenalina y ácido vanililmandélico). Los estudios radiológicos son los habituales para los tumores abdominales: ultrasonidos, TAC y RMN.
La adrenalectomía está indicada cuando se detecte feocromocitoma, pero dada la tan baja incidencia de feocromocitomas malignos, actualmente se realiza cirugía conservadora en la medida de lo posible. En los últimos años, diferentes grupos de estudio de la enfermedad han comenzado a utilizar la resección laparoscópica con buenos resultados (especialmente útil en tumores de pequeño tamaño, detectados en fase asintomática).
5. Páncreas.
Las lesiones pancreáticas también pueden ser la única manifestación, y pueden preceder a las demás. Son de dos tipos:
- Quistes pancreáticos y cistoadenomas serosos (se encuentran en alrededor del 70% de las autopsias). No suelen dar sintomatología. A menudo los quistes serosos son múltiples, pudiendo llegar a ocupar toda la glándula. Rara vez se hacen sintomáticos – al aumentar de tamaño- ocasionando complicaciones (dolor local, obstrucción del conducto biliar, pancreatitis e insuficiencia pancreática). Mientras sean asintomáticos, únicamente requieren control ecográfico periódico. Cuando aparece dolor puede recurrirse al drenaje percutáneo.
- Tumores de los islotes pancreáticos. Son tumores neuroendocrinos, más frecuentes en pacientes con feocromocitomas. Aparecen con una baja prevalencia, muchos de ellos de crecimiento lento. Pueden metastatizar en el hígado.
Una vez se han detectado quistes pancreáticos, es aconsejable realizar TAC o RMN como parte del estudio preliminar. Posteriormente se puede utilizar la ecografía para los controles periódicos de los mismos.
6. Tumor de saco endolinfático.
Ha sido recientemente cuando se ha identificado este tipo de tumor como integrante del síndrome que conforma la enfermedad. El paciente acude a la consulta manifestando una pérdida auditiva, acompañada de tinnitus y en ocasiones vértigo. Otras veces aqueja cambios en la textura del sonido, sin saber matizar las características de lo que percibe como anormal. También pueden aparecer síntomas relacionados con estructuras del ángulo ponto-cerebeloso. El crecimiento del tumor es lento, pero altamente destructivo.
El diagnóstico se basa en la sospecha clínica, y en la RMN de canal auditivo interno. El diagnóstico diferencial ha de realizarse con el adenoma del oído medio. Aunque hasta ahora no se han descrito casos de metástasis, y el crecimiento del tumor es lento, está indicada la monitorización clínica -con controles audiométricos- y radiológica. La detección precoz es crucial, pues la cirugía puede prevenir la progresión de los síntomas.
La prevalencia que se ha estimado para este tipo de tumores es de un 11%.
7. Epidídimo: quistes y cistoadenomas.
En el epidídimo se pueden encontrar quistes simples y cistoadenomas, uni o bilaterales. Los quistes suelen ser asintomáticos. Los cistoadenomas son tumores benignos, y particularmente los cistoadenomas papilares son típicos de esta enfermedad. Pueden aparecer en la adolescencia. Los tumores bilaterales pueden ocasionar oligozoospermia, disminución del volumen de eyaculado, obstrucción de los conductos seminíferos, y cosecuentemente problemas de fertilidad. Se detectan mediante exploración manual y ultrasonidos. La cirugía puede estar indicada para aliviar las molestias.
8. Tumor papilar anexal de probable origen mesonéfrico.
Es el equivalente al cistoadenoma epididimario, e histológicamente similar. Aparece en el ligamento ancho, a lo largo de las paredes uterinas o en la parte posterior de la vagina. Podría tratarse de una manifestación patognomónica de la enfermedad, pero hace poco tiempo que se ha comenzado a informar y registrar este tipo de tumores.
Puede presentarse con dolor hipogástrico, aunque muchas veces son asintomáticos, y se detectan durante exploraciones de rutina. Algunas de estas pacientes han sido histerectomizadas por sospecha inicial de carcinoma.
Cualquiera de los tumores descritos puede surgir a lo largo de la vida del individuo, aunque algún tipo de mutación del gen se asocie con mayor frecuencia a un tipo concreto de tumores. El diagnóstico precoz es el objetivo fundamental del seguimiento de estos pacientes, por lo que los controles periódicos deben incluir todos los órganos en riesgo.
Muchas otras lesiones han sido descritas con mayor o menor frecuencia en los enfermos de von Hippel Lindau (ependimomas, astrocitomas, meningiomas, carcinoma hepatocelular, quistes pulmonares, tumor de células germinales, carcinoma medular de tiroides, quistes óseos, nevus y manchas café con leche, etc.), aunque probablemente en muchos casos se trate de hallazgos sin asociación con la enfermedad.
El mayor problema al que se enfrentan actualmente estos pacientes es encontrar un médico que conozca la enfermedad. Todo lo visto hasta ahora nos muestra que el diagnóstico precoz es esencial para la supervivencia, y de ahí la importancia del buen conocimiento de los síntomas que el paciente refiere.
Aunque el seguimiento de la enfermedad debería llevarse a cabo coordinando las diferentes especialidades, habitualmente siguen revisiones en un solo servicio, el de la patología que estableció el diagnóstico. En la mayoría de los casos no se ha realizado el estudio genético, ni se ha elaborado el árbol genealógico familiar.
Texto con información para profesionales de la salud, preparado por la Dra. Karina Villar.








Yo estoy diagnosticada con VHl, al leer este documento,es como si estubiera contando mi historia.Mi hija mayor falleció ,supuestamente por un feocromocitoma,no fue estudiada,mi otra hijita ya laOperaron de las 2 suprarrenales, yo soy operada bilateral y hoy mis riñones están con tumores,quistes en el Páncreas,tumor en el saco endolinfatico,tumor en el cono medular,miomas de 15cm en el útero ,nódulos en las Mamas etc,etc,..un urólogo sugere que me saque los riñones y seguir en Diálisis de por vida…prefiero morir,quiero vivir lo que me queda de vida,no estar día por medio en una camilla dialisandome,me iré feliz. A los 22 años fui por unos análisis y hoy a los 51 años ya no quisiera mas Resonancias.atte. Victoria. Chile
Hola Graciela:
Teneis una asociación en Argentina para pacientes de Von Hippel Lindau.
Carlos Alberto Fredes / Presidente (AAF-VHL)
Asociación Argentina de Familias de Von Hippel-Lindau (AAF-VHL)
Calle: 70 Nº 3566 Ciudad de Necochea Cód. Pos. (7630)
Provincia de Buenos Aires – República Argentina
asociacion.arg.vhl@hotmail.com
Un Saludo y espero haber contribuido a ayudarte.
Hola tengo una amiga que el hijo padece de esta enfermedad (VHL) y necesitaría saber si existe alguna fundación o asociación civil de apoyo a familiares y de actualización sobre los avances positivos que ha detectado la ciencia, ya que ésta permanentemente avanza gracias a Dios.
Hace dos meses y medio falleció el papá (53 años) del chico (aproximadamente 25 años) Les pido por favor ayuda para contener a la madre y como pueden descartar que el otro hijo (20 años) no padezca también de VHL.
Muchas Gracias, no se olviden por favor, somos de la ciudad de La Plata, Provincia de Buenos Aires, Argentina.